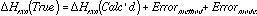
Division of Polymer Chemistry
Symposium on Stability of Plastics and Rubbers
American Chemical Society
San Diego Meeting
March, 2001
**Draft**
For Publication in Polymer Preprints
Volume 42, No. 1
March, 2001
A COMBINED MOMO ANALYSIS OF THE ONSET OF THERMAL DEGRADATION IN POLYMERS
Ernest Chamot
Chamot Laboratories, Inc.
530 E. Hillside Rd.
Naperville, Illinois 60540
Abstract
In a strictly thermal process, the thermal degradation of polymers by a free radical mechanism is initiated by a homolytic bond dissociation. This provides the initial source of free radicals which can undergo subsequent beta-scission, rearrangement, elimination, H-abstraction reactions, etc. that break the polymer down. By analyzing the Bond Dissociation Energy (BDE) of each bond in the polymer, the inherent limitations to thermal stability can be identified. A combined Molecular Orbital-Molecular Orbital (MOMO) method has been developed to accurately calculate individual, absolute BDE's for polymers, based on BLYP Density Functional and AM1 semiempirical calculations. This method provides energies accurate enough to analyze the effect of polymerization irregularities, and to predict thermal decomposition temperatures. The development and application of this method to the commercialization of a polyolefin process and the solution of a "wear" problem in the furniture industry will be discussed.
For the analysis of BDE's in polymers, the difference between the corresponding BLYP and AM1 calculations on a small model compound were used to estimate the systematic error due to the AM1 method in calculating the BDE in the polymer:
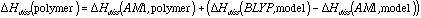
This is analogous to the general empirical corrections for ab initio BDE's developed by Irikura,10 but is more specific in that a correction is calculated for each individual bond.
A set of polymer models was developed to realistically represent each of the major components and each of the bond types in the polymers: a series of propylene and butylene octamers, an MFR trimer, a UFR tetramer, an alpha-D-glucose dimer (to represent cellulose), and a coniferyl alcohol pentamer (to represent lignin). These were modeled at the AM1 semiempirical level. Then a corresponding small model compound was modeled at both the AM1 and BLYP Density Functional level, and an AM1 to BLYP correction calculated. The small model compounds needed to include atoms at least 3 bonds away, and to consist of 10-29 heavy atoms for the corrections to converge. Correction factors for 36 different types of bond have been calculated. These corrections were then applied to the results of semiempirical AM1 calculations on the polymer model compounds, to extrapolate BLYP quality BDE's.
Results and Discussion
In the case of polyolefins, all of the backbone bonds are very similar (saturated C-C single bonds) so that the AM1 BDE errors, although large, are systematic: the same trends are observed with or without the AM1 to BLYP correction. Analysis of stereoregular isotactic PolyPropylene (i-PP) lead to the interesting prediction that, due to the alternating anti-gauche conformation of the backbone bonds in forming the known, 3-1 helical crystal structure,11 i-PP should have alternating strong and weak bonds, differing by about 2.5 kcal. This could explain the observation of "oligomers" in Mass Spectral analysis of i-PP, even when the low MW oligomers have been extracted: the i-PP may just be decomposing thermally in even monomer units.
The effect of various irregularities in i-PP (monomer inversion, monomer reversal, head-to-head or tail-to-tail coupling, 1,3-insertion, etc.) on BDE's was analyzed with the MOMO method, to determine their effect on the inherent thermal stability of polypropylene. The weakest link in a stereoregular i-PP is stronger than in propylene polymers with irregularities, but an occasional tacticity irregularity doesn't significantly weaken the chain (70.9 vs. 71.2 kcal). Orientation irregularities, however, can drop the stability by 3-5 kcal/mole. A head-to-head junction, either from a coupling reaction or from a monomer reversal, would result in a BDE of 67.8 kcal (vs. 71.2 kcal). A tail-to-tail junction would be more stable (75.6 kcal), but this would not increase the thermal stability of the polymer, since it would still be limited by the BDE of the weakest link.
A similar analysis of the bonds in polybutenes predicts that even a stereoregular, alternating i-butene/n-butene copolymer would be less stable than propylene, even with the least stable irregularity (64.2 kcal vs. 67.8 kcal). Any i-butene/i-butene combination would introduce an even weaker link in the backbone by 2-3 kcal, so it is clear that viscous polybutene is inherently less stable than i-PP.
To analyze the stability of particle board resins with respect to the High Temperature Corrosion (HTC) problem, it was desirable to relate the stability to decomposition temperatures, as well as to determine the structural features responsible for limiting thermal stability. Accordingly, the corrected BDE for each bond in each component was used to estimate the free energy of bond dissociation, �Gdiss, by combining it with entropies calculated from the AM1 frequency calculations. Simple Transition State Theory turned out to be sufficient for predicting rate constants, by using this estimate as an approximation of the energy barrier in the Arrhenius equation. A typical unimolecular value of 1016 sec-1 was used for the preexponential factor, A, and 1st order rate constants were calculated for each reaction (at 500° C for example) via:

Decomposition temperatures were calculated, by deriving the equation for a half-life from the integrated rate law for a 1st order reaction, and solving for the temperature at which the half-life is equal to the time scale of the process. Decomposition temperatures calculated on the Thermal Gravimetric Analysis (TGA) timescale (50° C/min temperature rise) reproduced experimental TGA spectra of mixed Melamine-Formaldehyde Resin (MFR) and Urea-Formaldehyde Resin (UFR) very well: 282° C and 335° C calculated vs. the observed broad 250-350° C decomposition peak. Calculated decomposition temperatures on the cutting contact timescale (0.2 msec) are consistent with frictional temperatures from finite element estimates of up to 450° C. This tends to confirm the validity of these calculations.
The weakest bond in the MFR model compound that is predicted to limit its stability is the C-O bond of a butyl ether. This group is not inherently part of a Melamine-Formaldehyde Resin. It is an artifact of the use of butyl alcohol as a process aid. Residual butyl alcohol is known to sometimes remain incorporated as butyl ether. These calculations predict that whenever butyl ether residues are present, their C-O bond will be the weak link that limits the thermal stability of MFR. This explains the variability in the thermal stability of MFR coatings: relatively small amounts of butyl alcohol residues will significantly reduce the thermal stability of MFR and decomposition will result in HTC.
One impractical solution would be to avoid the use of butyl alcohol in the MFR process, but by understanding the decomposition mechanism, another solution presented itself. A special grade of MFR is made with a process called "hardening," by prolonged heating with an acid catalyst. This forces additional condensation and crosslinking, but should also displace the residual butyl alcohol. This should result in a more thermally stable resin, which would in turn reduce the decomposition processes responsible for HTC. Subsequent wear experiments with "hardened" MFR samples confirmed this prediction.
Conclusion
Polymer thermal stability can be predicted based on calculated bond dissociation energies, relying on the fact that bond dissociation will determine the onset of thermal degradation, and that the weakest link in the chain limits the thermal stability of the entire molecule. A combined Molecular Orbital-Molecular Orbital method has been developed, using BLYP Density Functional calculations as the high level MO Theory for accuracy, and using AM1 semiempirical calculations as the lower level MO Theory, in order to model polymers with realistic model compounds. A set of correction factors was developed, relating AM1 to BLYP calculations. With BDE's calculated in this way, initial fragmentation patterns, relative polymer stabilities, and polymer thermal decomposition temperatures can be calculated that are consistent with experiment.
Stereoregular isotactic polypropylene is predicted to have alternating strong and weak backbone bonds, and the irregularity that would most reduce thermal stability is formation of a head-to-head junction. Viscous polybutenes are inherently less stable than polypropylene. The initial stages of the High Temperature Corrosion mechanism have been identified, and a counter intuitive solution to HTC has been found.
1 Minsker, K.S.; Kolesov, S.V.; Yanborisov, V.M. Polymer Deg. and Stab. 1984, 9, 103.
2 Zubkov, V.A.; Bogdanova, S.E.; Yakimansky, A.V.; Kudryavtsev, V.V. Macromol. Theory Simul. 1995, 4, 209.
3 Munk, P. Introduction to Macromolecular Science, John Wiley & Sons, New York, 1989, p 212.
4 Stewart, H.A. Forest Products Journal 1989, 3, 25.
5 Chamot, E.; Rollin, A.; Schoenberg, M.; Firth, B. "Probing Polyolefin Stabilities by Semiempirical Methods," presented at the Central Region ACS Meeting, Akron, Ohio, May 31-June 2, 1995.
6 Noland, M.; Coitino, E.; Truhlar, D. J. Phys. Chem. A 1997, 101(7), 1193.
7 Jursic, B. THEOCHEM 1996, 366(1-2), 103; Jursic, B.; Martin, R. Int. J. Quantum. Chem. 1996, 59(6), 495.
8 Cioslowski, J.; Liu, G.; Moncrieff, D. J. Amer. Chem. Soc. 1997, 119(47), 11452.
9 Humbel, S.; Siever, S.; Morokuma, K. J. Chem. Phys. 1996, 105, 1959.
10 Irikura, K.K. J. Phys. Chem. A 1998, 102, 9031.
11 Crawford, J.W. J. Soc. Chem. Ind. 1949, 68, 201.
